
La evidencia clínica obtenida a partir de estudios realizados en hermanos sugiere que la intervención temprana ofrece múltiples oportunidades para mejorar los resultados en pacientes a través de un tratamiento específico de la enfermedad y el inicio temprano de TRE, si se encuentra disponible.1-6
Se ha demostrado que la TRE, independientemente del momento del inicio, mejora los parámetros clínicos claves tales como mediciones de resistencia y pulmonares, que son críticas para la calidad de vida, el mantenimiento de la ambulación y las actividades de la vida diaria.7,8
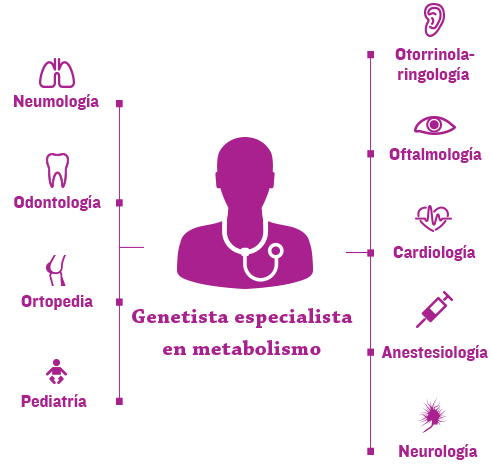
La nueva era de cuidado de enfermedades genéticas, complejas y progresivas, tales como mucopolisacaridosis (MPS), depende de la coordinación eficiente del equipo de salud de cada paciente a través de un hogar médico.1
Normalmente los genetistas y/o especialistas del metabolismo se encuentran en el centro del hogar médico y ayudan a coordinar la atención multidisciplinaria y un plan de tratamiento personalizado.2,3
Los cardiólogos tienen un rol fundamental en la atención coordinada multidisciplinaria de los pacientes con MPS por varias razones2,4:
Dada la prevalencia y gravedad del compromiso cardíaco, la evaluación cardíaca continua es un componente clave del tratamiento efectivo en estos pacientes.3,4 Las manifestaciones cardíacas más comunes de MPS que requieren monitoreo incluyen las siguientes5:
Muchas enfermedades MPS cuentan con lineamientos de atención disponibles y recomendaciones consensuadas específicas de la especialidad con respecto a la atención de por vida de MPS. Generalmente, los lineamientos recomiendan3,6:
Las evaluaciones tempranas y continuas de un equipo de salud coordinado pueden mejorar los resultados de los pacientes, a su vez pueden ayudar a prevenir daños irreversibles.6
A pesar de que el compromiso cardíaco se observa en todos los subtipos de MPSs la mayoría de los estudios demuestra que tiende a ocurrir de manera más temprana y frecuente en MPS I, II, y VI que en MPS III y IV. Sin embargo, es importante recordar que los signos y síntomas son impredecibles, clínicamente heterogéneos tanto para los distintos subtipos de MPSs, como dentro de cada uno de ellos. Los pacientes pueden presentar signos cardíacos clásicos y no clásicos de MPSs así como también una enfermedad de progresión lenta o rápida.4 Esto subraya la importancia del monitoreo cardíaco basal y constante en todos los pacientes con MPS.3
Existe una cantidad de revisiones de expertos que establecen las últimas recomendaciones para la evaluación y el monitoreo de anomalías cardíacas en pacientes con MPS.3,7-9 Para muchos de los subtipos de MPSs también existen guías de tratamiento y recomendaciones consensuadas específicas de cardiología con respecto a la atención crónica.3,6,10
Estas revisiones y guías incluyen las siguientes recomendaciones generales3,6,10:
La figura a continuación detalla las técnicas de diagnóstico y exámenes recomendados para evaluar la anatomía y función cardíaca en pacientes con MPS tanto en el diagnóstico inicial como en intervalos regulares posteriores. Los intervalos de evaluación recomendados oscilan entre 1 a 3 años, según el subtipo de MPS.4

Se deben considerar evaluaciones cardíacas de rutina adicionales antes de intervenciones de cirugía mayor, incluyendo un auscultamiento minucioso del corazón y pulmones, así como también mediciones de presión arterial en el brazo y pierna derechos.4
Los cardiólogos implicados en la atención continua de pacientes con MPS siempre deben recordar lo siguiente4:
La frecuencia de las evaluaciones y participación de especialistas específicos varía según los distintos tipos de MPS. En pacientes con enfermedades MPS asociadas con complicaciones primarias neurodegenerativas y cognitivas, tales como MPS I, II, y III, se recomiendan evaluaciones psiquiátricas y de comportamiento neurológico adicionales.6,11,12
Además de las evaluaciones específicas de la especialidad que deben realizarse para favorecer resultados positivos a largo plazo en pacientes con MPS, el médico coordinador, normalmente el genetista y/o especialista del metabolismo, puede realizar algunas acciones importantes con relación a la salud en general. Su rol en educar a otros profesionales de la salud (ej, dentistas, fisioterapeutas, pediatras, médicos de familia), a las familias sobre la enfermedad y las estrategias generales de cuidado es crítico y debe incluir lo siguiente:
Tanto las evaluaciones específicas de la enfermedad como los exámenes físicos e intervenciones generales de salud deben seguir los lineamientos recomendados, que pueden variar según el subtipo de MPS.3
Los avances en el tratamiento de enfermedades MPS favorecen los resultados a largo plazo en pacientes, por lo que se necesitan nuevos enfoques de atención de por vida.
Con el paso de los años, algunos pacientes aprenden a manejar su propia salud, esto hace que la transición del cuidado del médico al escenario adulto sea crítica.3 Los médicos deben asegurar lo siguiente:
La transición de la atención pediátrica a la adulta y la atención a largo plazo son áreas fundamentales a tener en cuenta en los planes de atención de pacientes adolescentes y adultos.3 Las consideraciones a largo plazo son mejor abordadas en un centro con amplia experiencia en MPS, y requieren una coordinación minuciosa entre las especialidades.3,14 Las cuestiones a largo plazo incluyen, pero no se limitan a las siguientes:
La atención a largo plazo de enfermedades MPS—incluyendo evaluaciones continuas y estrategias de transición específica del centro desde la atención pediátrica hasta la adulta— puede llevar a una mejora considerable en la calidad de vida y a un mejor futuro para los pacientes.3,14-16
Debido a que las manifestaciones clínicas de enfermedades mucopolisacaridosis (MPS) son multisistémicas, se requiere un enfoque multidisciplinario y específico del paciente, para reconocer y manejar las complicaciones de manera proactiva. La participación del cardiólogo en este proceso es fundamental ya que las cirugías cardíacas son frecuentes en pacientes con MPS.1-4
Los pacientes con enfermedades MPS normalmente atraviesan una cantidad de cirugías durante sus vidas. Un estudio de evolución natural que evaluó un cohorte de 325 pacientes con Morquio A (MPS IVA) halló que más del 70% de los pacientes fue sometido por lo menos a una cirugía.5

Los pacientes con MPS tienen una tasa alta de mortalidad perioperatoria debido a múltiples factores, incluyendo la obstrucción de las vías superiores e inferiores, inestabilidad cervical espinal, insuficiencia respiratoria, morbilidad cardiovascular, e infecciones frecuentes.3,5,6 Por ejemplo, las complicaciones quirúrgicas resultaron en una tasa de mortalidad del 11% en pacientes con Morquio A (n=27).7
Es fundamental crear un plan quirúrgico que involucre a un equipo multidisciplinario de especialistas que también tengan experiencia, preferentemente, en el manejo de pacientes con MPS.6
La atención cardiológica del paciente quirúrgico suele incluir el tratamiento quirúrgico de diversas manifestaciones. Entre los procedimientos cardíacos exitosos documentados en pacientes con MPS, encontramos4:
La evaluación del riesgo quirúrgico y el monitoreo perioperatorio son componentes fundamentales de un plan quirúrgico personalizado, pudiendo también reducir el riesgo de resultados adversos de la cirugía y de muerte en pacientes con MPS.6,10,11


References: 1. McGill JJ, Inwood AC, Coman DJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age—a sibling control study. Clin Genet. 2010;77(5):492-498. doi:10.1111/j.1399-0004.2009.01324.x. 2. Furujo M, Kubo T, Kosuga M, Okuyama T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab. 2011;104(4):597-602. doi:10.1016/j.ymgme.2011.08.029. 3. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13-18. 4. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 5. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 6. Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95-101. doi:10.1097/GIM.0b013e3181fea459. 7. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 8. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63-72. doi:10.1016/j.ymgme.2013.11.015. 9. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 10. Bagewadi S, Roberts J, Mercer J, Jones S, Stephenson J, Wraith JE. Home treatment with Elaprase® and Naglazyme® is safe in patients with mucopolysaccharidoses types II and VI, respectively. J Inherit Metab Dis. 2008;31(6):733-737. doi:10.1007/s10545-008-0980-0. 11. BioMarin Pharmaceutical Inc. VIMIZIM website. http://www.vimizim.com/. Accessed December 21, 2015. 12. BioMarin Pharmaceutical Inc. NAGLAZYME website. http://www.naglazyme.com/. Accessed December 21, 2015. 13. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416.
References: 1. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 3. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 4. Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183-1197. doi:10.1007/s10545-011-9359-8. 5. Mohan UR, Hay AA, Cleary MA, Wraith JE, Patel RG. Cardiovascular changes in children with mucopolysaccharide disorders. Acta Paediatr. 2002;91(7):799-804. 6. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416. 7. Solanki GA, Martin KW, Theroux MC, et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36(2):339-355. doi:10.1007/s10545-013-9586-2. 8. Zafeiriou DI, Batzios SP. Brain and spinal MR imaging findings in mucopolysaccharidoses: a review. AJNR Am J Neuroradiol. 2013;34(1):5-13. doi:10.3174/ajnr.A2832. 9. Lachman R, Martin KW, Castro S, Basto MA, Adams A, Teles EL. Radiologic and neuroradiologic findings in the mucopolysaccharidoses. J Pediatr Rehabil Med. 2010;3(2):109-118. doi:10.3233/PRM-2010-0115. 10. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405-418. doi:10.1542/peds.2006-2184. 11. Neufeld EF, Muenzer J. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, et al, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3421-3452. 12. Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi:10.1186/1750-1172-6-72. 13. James A, Hendriksz CJ, Addison O. The oral health needs of children, adolescents and young adults affected by a mucopolysaccharide disorder. JIMD Rep. 2012;2:51-58. doi:10.1007/8904_2011_46. 14. Coutinho MF, Lacerda L, Alves S. Glycosaminoglycan storage disorders: a review. Biochem Res Int. 2012;2012:471325. doi:10.1155/2012/471325. 15. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of child neurology. New York, NY: McGraw-Hill; 1996:1141-1166. 16. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48.
References: 1. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27-S34. 2. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29. doi:10.1542/peds.2008-0416. 3. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 4. Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183-1197. doi:10.1007/s10545-011-9359-8. 5. Harmatz P, Mengel KE, Giugliani R, et al. The Morquio A clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54-61. doi:10.1016/j.ymgme.2013.01.021. 6. Walker R, Belani KG, Braunlin EA, et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211-219. doi:10.1007/s10545-012-9563-1. 7. Lavery C, Hendriksz C. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59-66. doi:10.1007/8904_2014_298. 8. Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22(9):901-907. doi:10.1111/j.1460-9592.2012.03904.x. 9. Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi:10.1186/1750-1172-6-72. 10. Solanki GA, Martin KW, Theroux MC, et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36(2):339-355. doi:10.1007/s10545-013-9586-2. 11. Vitale MG, Skaggs DL, Pace GI, et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333-339. doi:10.1016/j.jspd.2014.05.003. 12. Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012;107:15-24. doi:10.1016/j.ymgme.2012.07.018. 13. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983.


